EXERCISE 5
Exercise 5: Running WRF-Chem with cycled chemistry
***************************************************************************
Purpose: To familiarize the user with
the use of the WRF-Chem model with one of the most popular settings
- cycling the chemistry from
a previous forecast. When making a new forecast there will be (should be)
fresh atmospheric observations to use for the analysis fields.
Unfortunately, the state of the atmospheric chemistry is not as well
observed. Therefore, one must be creative in the methodology to
accurately describe the initial state of the
chemical structure of the atmosphere.
In the WRF-Chem model the atmospheric chemistry can be initialized using
an idealized vertical profile (section 4.5 of the User's Guide). But once a
forecast has been made the tropospheric chemistry is probably more accurately
described by a WRF-Chem model output than the idealized profile. Therefore, one
can use a previous chemistry forecast along with the meteorological analysis
to produce the most realistic initial state for starting a new forecast. To
accomplish this, a previous chemistry forecast is read in through
the auxiliary ports and output by the real.exe program.
***************************************************************************
Note: The assumption is that the user
is very familiar with the set-up and running of the WRF
model and has run the WRF-Chem model. You will need to have
access to the WRF-Chem User's guide and quick_start guide to
complete this exercise and understand why it worked. Very little
details are provided in this assignment due to the wealth of
information in the other available resources.
- Be sure to have your environmental
variables set and move to your local directory.
working in /kumquat/users/${USER}
- Start with the simulation results from tutorial exercise 4, or 3.
The model should have made at least a 12 forecast and have produced a new wrfout data file at
the new forecast time (12 hours in this case). That is, set in your namelist.input file
history_interval = 60, 60, 60,
frames_per_outfile = 12, 12, 12,
If you have only one wrfout file, then repeat the simulation so that at 12 hours you have a new wrf output file.
wrfout_d01_2010-07-14_00:00:00 <- hours 0 to 11
wrfout_d01_2010-07-14_12:00:00 <- hour 12
The reason being the cycling routine assume the first time in the input file is the forecast start time.
- Save the output from the previous 12 forecast (most likely exercise 3, but
it could be another) to a different directory. For reference it is assumed that the previous forecast
should have started at 00 UTC on 14 July 2010.
mkdir Output
mv wrfout_d01_2010-07-14_00:00:00 Output
mv wrfout_d01_2010-07-14_12:00:00 Output
etc.
- It is assumed you still have the WPS output file (the "met_em" files) in your WRFV3/test/em_real directory.
If not, follow the steps provided in exercise_1 to produce the met_em files and then continue this exercise.
- Link the saved 12 hour forecast output file to your run directory and name the input file "wrf_chem_input_d01"
ln -sf Output/wrfout_d01_2010-07-14_12:00:00 wrf_chem_input_d01
- Next set the correct forecast start date, turn on auxiliary input 12 and set chem_in_opt=1 in the
namelist.input file. Remember the auxiliary input file ports are set-up for different input needs.
auxinput_5 -> Anthropogenic emissions
auxinput_6 -> Biogenic emissions (BEIS, MEGAN)
auxinput_7 -> Surface biomass burning fields
auxinput_8 -> GOCART background fields
auxinput_12 -> Chemistry initial fields
auxinput_13 -> Volcanic ash emissions
auxinput_14 -> Aircraft emissions
auxinput_15 -> Green House Gas emissions
For this exercise one only needs a 1 hour forecast, but you can leave the forecast run time at 12 hours long
and terminate the run when you like. Just be sure that the forecast is
starting at 12 UTC and not 00 UTC on 14 July 2010. The namelist.input file should have set:
run_hours = 12,
start_day = 14, 14, 24,
start_hour = 12, 00, 12,
...
end_day = 15, 16, 25,
end_hour = 00, 00, 12,
...
auxinput12_inname = 'wrf_chem_input',
io_form_auxinput12 = 2,
...
chem_in_opt = 1,
...
- Now run real.exe to produce the input file the next forecast. Examine the text output from running real.exe (the rsl.out.0000 file).
The file should contian the following information about 30 lines down from the top.
INITIALIZING CHEMISTRY WITH OLD SIMULATION
mediation_integrate: med_read_wrf_chem_input: Open file wrf_chem_input_d01
mediation_integrate: med_read_wrf_chem_input: Read chemistry from wrfout at time 2010-07-14_12:00:00
...
- Examine the wrfinput file as well as the wrf_chem_input_d01 file as well using ncview (or similar utility). Look at fields like
CO in both. The old 12 hour forecast (wrf_chem_input_d01) and the wrfinput_d01 file should be identical. That is, the CO field in the
wrf_chem_input_d01 file should look like locations of major cities and have a range from 0.06 to 0.15 ppmv and the wrfinput file
is the same. IF the wrfinput_d01 file has CO that ranges from 0.06 to 0.08 and is horizontally uniform you have an error.
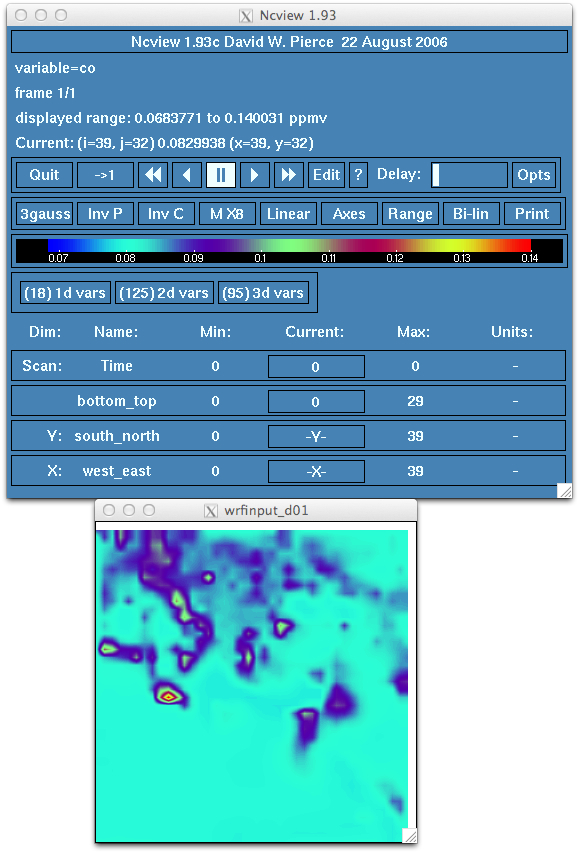
- Run wrf.exe for the forecast and verify that the model starts with the cycled chemistry. The initial time in the wrfout file
should look like the wrfinput file, but evolve from the starting point.
Did you get an error asking for the wrfchemi_12z_d01 emissions
data file? When using RETRO/EDGAR emissions the emitted species are updated monthly so your wrfchemi_d01 file can be used for whenever your run
starts - 12 UTC emissions input in this example. If you obtain hourly emissions, then the 00 and 12 UTC files should both contain 12 hours of emissions data
(data from 00 to 11 UTC in the 00z data file and hours 12 to 23 UTC in the 12z data file).
So how do you fix the error? Simple, just get the emissions data for the day and time you are running and place it in the wrfchemi_12z_d01 file. This run looks for the
wrfchemi_12z_d01 data file as it is starting at 12z. In
this case, just copy the RETRO/EDGAR emissions file (wrfchemi_d01) to wrfchemi_12z_d01 and run wrf.exe again. Why wrfchemi_12z_d01 this time and wrfchemi_00z_d01 for
the earlier examples? It is because the run starts at 12 UTC and it needs a file containing emissions from 12 and 23 UTC. The previous runs started at 00 UTC and so
they needed an emissions data set with times between 00 and 11 UTC. This is true even if the emissions are read in just once for the entire run.
Note: Baroclinic waves typically take 3 to 4 days to cross a coarse simulation domain (3000 km x 2000 km for example) so cycle your WRF-Chem run several days before
accepting the simulation results. This will give enough time for emissions and vertical transport to spin-up a reasonably looking
chemistry field in the lower troposphere.
This concludes WRF-Chem tutorial exercise 5
To compare solutions, download desired files from here.
Return to WRF-Chem Tutorial Page